Chapter 19: Berg, Cohen and Boyer Invent Recombinant DNA and Gene Cloning
“The general procedure described here is potentially useful for insertion of specific sequences from prokaryotic or eukaryotic chromosomes or extrachromosomal DNA into independently replicating bacterial plasmids.”
The first of the three technologies that revolutionized molecular biology is gene cloning (also commonly referred to as DNA cloning or molecular cloning). This story, which builds on the discovery of restriction endonucleases, begins with the achievement of artificially joining unrelated DNA fragments together.
Creating recombinant DNA. Most of the credit for creating recombinant DNA went to Paul Berg (1926-2023), a member of the Biochemistry Department at Stanford University. Berg obtained his PhD from Case Western University in 1952. He did a postdoc in Copenhagen and later a postdoc with Arthur Kornberg at Washington University, where he joined the faculty in 1955. Berg then moved to Stanford in 1959 with Kornberg when Kornberg became head of Stanford’s Biochemistry Department. Berg won the Nobel Prize in 1980 for the invention of recombinant DNA (sharing the Prize with Walter Gilbert and Fred Sanger who were recognized for the invention of DNA sequencing, chapter 20.)
In his 1972 publication, Berg and postdoctoral fellows Jackson and Symons reported the joining Simion Virus 40 DNA (SV40) to the DNA of a derivative phage λ DNA (“λdvgal” which we will introduce below). The strategy for doing so was to linearize the circular genome of SV40 with the restriction enzyme EcoR1, which had been discovered in the laboratory of Herb Boyer at the University of California, San Francisco. This E. coli restriction enzyme, which played a central role in the history of gene cloning, has a more stringent recognition sequence than that of Smith’s Hemophilus enzyme (HinDII), which we considered in chapter 18. Hence, it cleaves at fewer sites than the Hemophilus enzyme. And, indeed, SV40 has only a single EcoR1 recognition sequence. Therefore, Berg and his team could linearize SV40 by cleavage with EcoR1 without breaking it into multiple fragments.
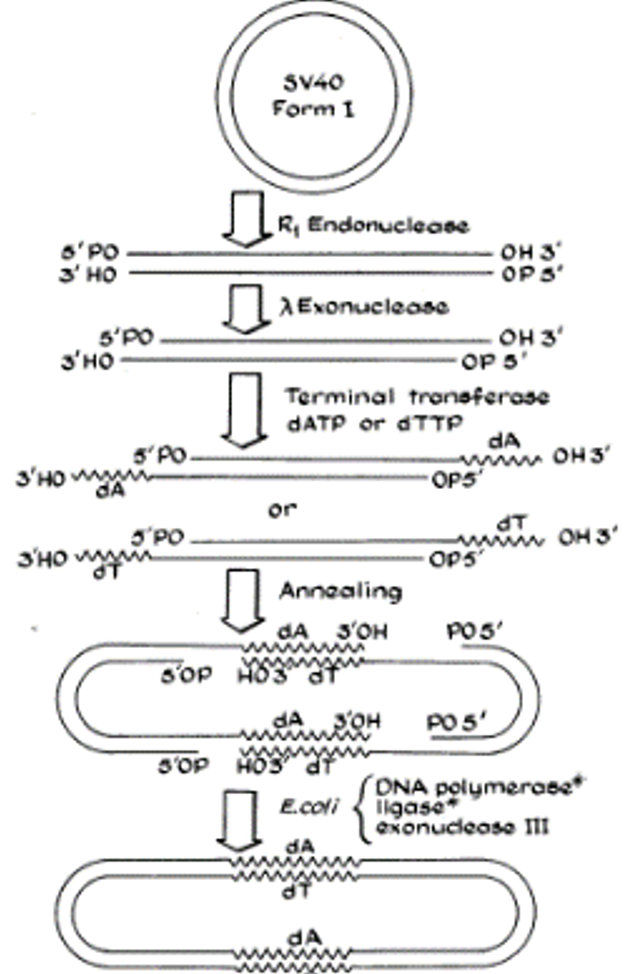
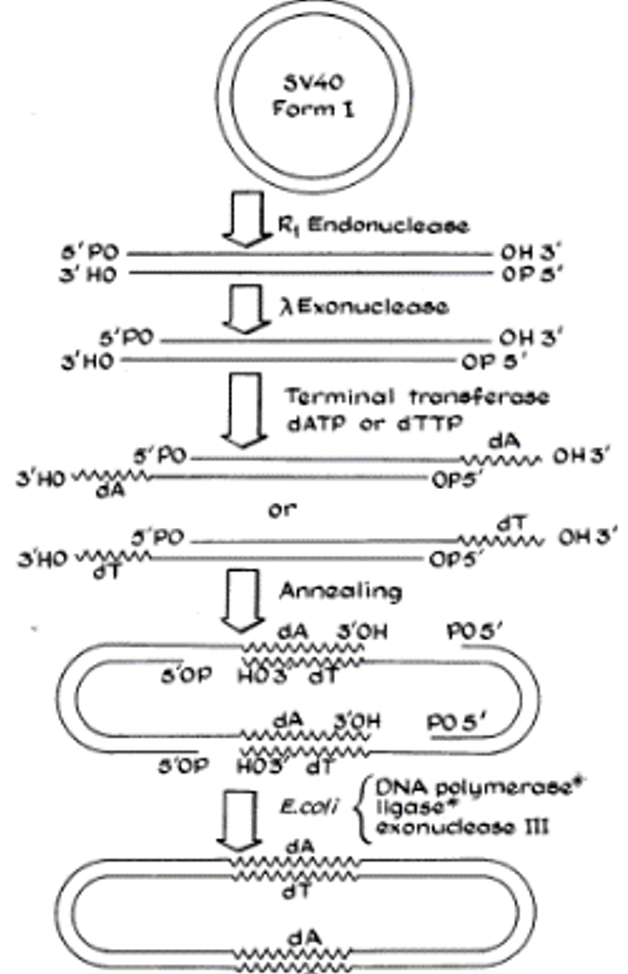
Figure 1 (below) from Berg and co-workers’ publication summarizes their strategy, using the dimerization of SV40 DNA as an illustration. SV40 was linearized by cleavage with EcoR1 (R1 in the figure). Next, after trimming back the 5’ ends with an exonuclease, a homopolymer of deoxy-T (dT) or deoxy-A (dA) was extended from the 3’ terminal ends using terminal transferase, an enzyme known to extend 5’ termini by creating homopolymers. This resulted in two kinds of linear DNAs, those with a poly-dT tail and those with a poly-dA tail. Because dT pairs with dA, the two linear DNAs would anneal to each other to create a circular DNA consisting of two molecules of SV40 DNA. Additional enzymatic treatments summarized at the bottom of the figure were used to create a covalently closed, circular molecule with two SV40 genomes fused to each other.
The stage was now set to create a recombinant DNA molecule. For this they used phage λ that had picked up genes for the galactose operon (which encodes enzymes that mediate metabolism of the sugar galactose) from the chromosome of E. coli in a natural process known as specialized transduction. Known as “λdvgal”, λ containing the galactose operon genes is capable of replicating as a plasmid in E. coli but lacks genes for propagation as a phage. Once again, EcoR1 was employed to generate a linear DNA of λ with gal genes. Using the strategy of Figure 1, Berg and co-workers joined linearized SV40 DNA with poly-dA tails to linearized λdvgal DNA to which had been appended poly-dT tails.
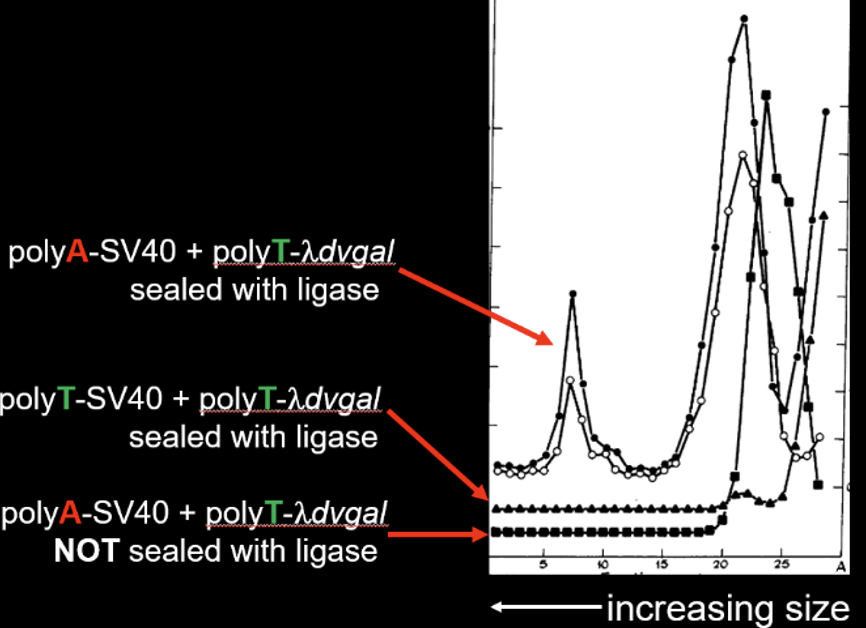
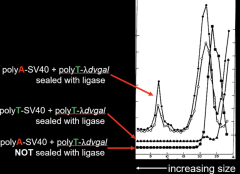
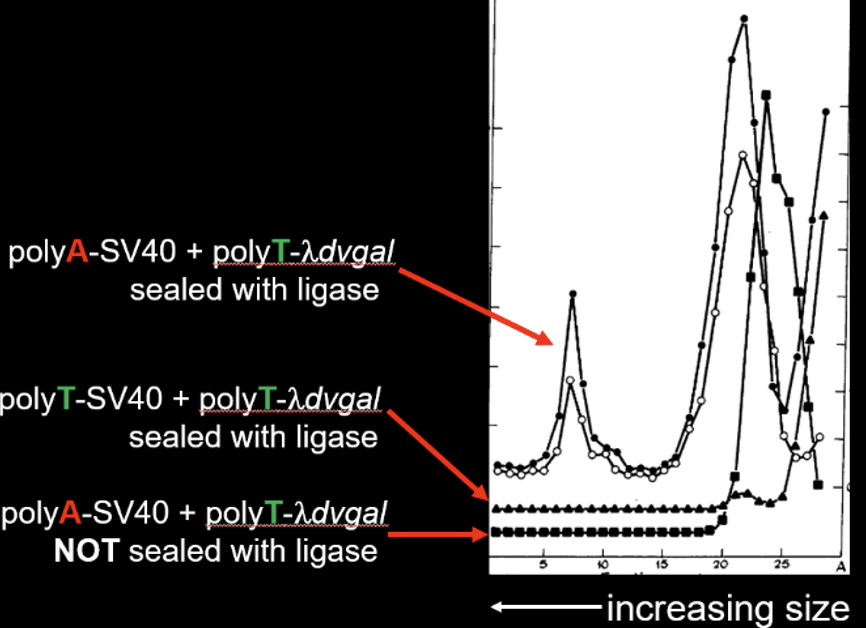
The experiment of Figure 3 helped demonstrate that they had covalently joined the two DNAs together. After the final ligation step, the DNAs were subjected to centrifugation through a sucrose gradient under alkaline conditions. At elevated pH, the two DNAs would not have been held together unless they had been covalently joined. As seen in the figure (labeling added) SV40 with a polyA tail that had been joined to λdvgal with a polyT tail exhibited a rapidly sedimenting peak in the gradient at about fraction 7. But if both DNAs had polyT tails, then no peak was observed. Also, if the two DNAs had not been treated with ligase, no peak was observed. Thus, Berg and co-workers had created for the first time in molecular biology a recombinant DNA molecule containing both mammalian and bacterial DNAs fused to each other.
But there is more to the story of recombinant DNA, and the history gets complicated! First, the SV40/λdvgal recombinant DNA was not introduced into E. coli. Hence, the SV40 DNA was not cloned in the sense that it was not amplified by propagation in the bacterium. Moreover, the site of cleavage by EcoR1 would have blocked the ability of λ to replicate as a plasmid. So, gene cloning had not yet been achieved and could not have been achieved with the λdvgal vector.
Second, the creation of recombinant DNAs triggered concerns that the unnatural joining of DNA molecules from different organisms (genetic engineering) could be a biohazard, inadvertently creating novel pathogens, for example. Indeed, Berg and others famously organized an international meeting in 1975 at the Conference Center at Asilomar State Beach in California to propose guidelines for the creation and use of recombinant DNA. Whether such fears were warranted is itself controversial. Few, if any, examples of biohazardous agents resulting from the creation of recombinant DNAs have emerged. Meanwhile, much fear surrounding the implementation of recombinant DNA technology has impeded its application for the betterment of humankind. For example, genetic engineering has been used to create a variety of rice known as Golden Rice that synthesizes a precursor of vitamin A. Such fortified rice could help address the prevalence of vitamin A deficiency in developing countries but its use as a food source has met with opposition from activists.
Third, who originated the idea of creating recombinant DNAs is controversial. Also in the Biochemistry Department at Stanford was Peter Lobban, a graduate student in the laboratory of Dale Kaiser, who had been working with λdvgal. Lobban proposed the idea of using the enzyme terminal transferase to create complementary sequences on the ends of two different DNA fragments for his doctoral thesis research so that they could be joined together. Also, he found that trimming the 5’ ends with an exonuclease made the ends a better substrate for terminal transferase, a finding that he shared with Berg. Lobban and Kaiser were slower to publish than Jackson, Symons and Berg, their report appearing the following year. In their 1973 publication, Lobban and Kaiser make it clear that they had developed a general method for creating recombinant DNAs: “The apparent generality of the method described here for of double-stranded DNA molecules to each other may provide a new way to study eukaryotic genes and their controlling elements. Any block of genes from any organism could be inserted into the genome of a temperate bacteriophage to generate a specialized transducing phage.” Nonetheless, and like Jackson, Symons and Berg, Lobban and Kaiser did not implement their recombinant DNA technology to clone a gene.
Berg in the video credits Lobban for independently developing the idea of creating recombinant DNA molecules using terminal transferase and for sharing the finding that exonuclease treatment facilitated its use.
EcoR1 generates sticky ends. Ironically, while the use of terminal transferase was being developed to create recombinant DNAs, three groups [Mertz and Davis (shown); Sgaramella; and Hedgpeth, Goodman and Boyer as reported in the same issue (November 15, 1972) of the Proceedings of the National Academy of Sciences] were making the surprising discovery that EcoR1 generates cohesive (“sticky”) ends (chapter 18) that can be joined to each other without the need for polynucleotide tails.
Thus, EcoR1-generated restriction fragments from any biological source would have identical, complementary 5’ overhangs that would anneal to each other. This meant, any two EcoR1-generated fragments could be recombined simply by base-pairing between their “sticky” ends followed by treatment with DNA ligase to seal the DNAs together.
Invention of gene cloning. The stage was now set for the revolutionary invention of gene cloning. The two heroes for this final part of the story are Stanley Cohen (1935-) and Herb Boyer (1936-). Cohen obtained an MD from the University of Pennsylvania School of Medicine in 1960, and then did a postdoc with the famous biochemist Jerald Horwitz at Albert Einstein College of Medicine in 1967. Cohen then joined the faculty of Stanford University in 1968 where he studied antibiotic-resistance plasmids and methods for introducing plasmids into E. coli. His partner in the invention of cloning, Boyer, obtained his PhD from the University of Pittsburg in 1963, did postdoctoral research at Yale University, and then joined the faculty of the University of California, San Francisco (UCSF) where he discovered EcoR1 and that it generates sticky ends as noted above. Boyer founded the first biotech company, Genentech in 1976 with venture capitalist Robert Swanson.
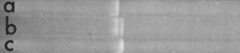
Cohen and Boyer started their collaboration in 1972 at a meeting in Hawaii. They famously had lunch at a delicatessen at Honolulu’s Waikiki Beach. Boyer, as we have seen, had discovered EcoR1 and that it generates sticky ends. Cohen, meanwhile, had developed a method for transforming E. coli with plasmid DNA. Unlike Streptococus and certain other competent bacteria (chapter 3), E. coli does not naturally take up naked DNA. However, Cohen had found that E. coli that was treated with calcium chloride was able to take up plasmid DNA and that cells harboring the plasmid could be selected by treatment with a plasmid-borne antibiotic resistance gene. One such plasmid, which carries a tetracycline resistance gene and is famously known as pSC101 (Stanley Cohen 101) harbors a single cleavage site for EcoR1. (The actual origin of pSC101 is uncertain as revealed in subsequent work by Cohen.) Importantly, Cohen and Boyer showed in their 1973 publication that EcoR1-cleaved pSC101 could be ligated to an unrelated, Salmonella plasmid (RSF1010) that also harbors a single EcoR1 cleavage site and carries a streptomycin-resistance gene. When E. coli was transformed with the two EcoR1-cleaved plasmids that had been ligated together, followed by selection for both tetracycline and streptomycin resistance, the transformants were found to harbor a new recombinant plasmid that consisted of both pSC101 and RSF1010. When this recombinant plasmid was treated with EcoR1 and subjected to electrophoresis, it was found to consist of two DNA fragments (b in the gel), one identical in size to EcoR1-cut pSC101 (a) and the other identical in size to EcoR1-cleaved RSF1010 (c).
Viola! The first recombinant DNA had been created and propagated in E. coli. Cohen and Boyer conclude their transformative (no pun intended) 1973 publication by noting, “The general procedure described here is potentially useful for insertion of specific sequences from prokaryotic or eukaryotic chromosomes or extrachromosomal DNA into independently replicating bacterial plasmids. The antibiotic resistance plasmid pSC101 constitutes a replicon of considerable potential usefulness for the selection of such constructed molecules, since its replication machinery and its tetracycline resistance gene are left intact after cleavage by the EcoRI endonuclease.”
In the video, Cohen and Boyer tell the history of their invention of gene cloning.
We conclude this chapter with this epilogue. Boyer teamed up with venture capitalist Robert Swanson to create the first biotechnology company, Genentech. Scientists at Genentech used the recombinant DNA technology to express genes in E. coli, famously producing human insulin as a therapeutic. Up until then insulin was obtained from the pancreas of pigs and cows. But now recombinant DNA technology had made it possible to produce the human hormone for treatment of diabetes.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}