Chapter 11: Pauling, Ingram and Sanger Link Genes to Proteins
“It immediately occurred to me that sickle-cell anemia must be a disease of the hemoglobin molecule, with sickle-cell hemoglobin having an abnormal structure...”
The underlying assumption of the Benzer and Crick et al. experiments that we have been considering is what came to be known as the “sequence hypothesis;” namely, that “the specificity of a piece of nucleic acid is expressed solely by the sequence of its bases, and that this sequence is a (simple) code for the amino acid sequence of a particular protein” as enunciated by Francis Crick in 1958 (chapter 14). Compelling evidence in support of the sequence hypothesis arose from studies of sickle cell anemia by Linus Pauling and Vernon Ingram and from insulin by Fred Sanger.
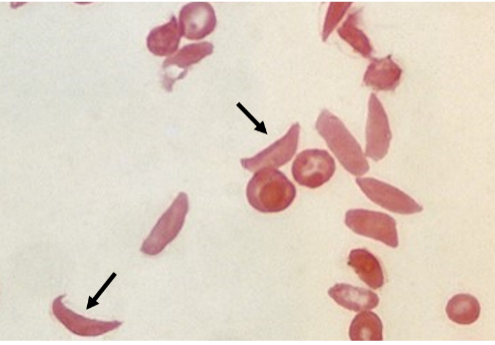
Sickle cell anemia is a genetic (inherited) disease of the β-globin gene carried on chromosome 11. (Hemoglobin consists of two α subunits and two β subunits.) The mutation is recessive and so the disease is only manifest in individuals who have inherited two copies of the mutant gene from their parents. Low oxygen levels in venous blood causes red blood cells to adopt a rigid, sickle-like shape, interfering with blood flow. It is most prevalent in Sub-Sharan Africa but also seen in India, the Arabian Peninsula and elsewhere. In the heterozygous state, that is, in individuals with mutant and wild type copies of the gene, it helps protect against malaria; the mutant β-globin interferes with the malaria parasite Plasmodium, which reproduces in red blood cells.
Pauling became intrigued by the disease while listening to a talk on sickle cell anemia by a physician from Boston. Pauling recalls that
{kind=link}
“I thought to myself, there are probably thousands of different chemical substances in the red blood cell, and I don't think I can understand why a cell would be twisted out of shape. Then, he [the speaker] went on to explain that they twisted out of shape in venous blood, but resume their normal flattened spherical shape in arterial blood. Within a few seconds the idea flashed through my mind that red cells contain a great amount of hemoglobin - about one-seventh of the content of a cell is hemoglobin molecules - and that the difference between venous blood and arterial blood is that in venous blood the hemoglobin is present as hemoglobin itself, whereas in arterial blood it becomes oxyhemoglobin because there are oxygen molecules attached to the iron atoms. .... It immediately occurred to me that sickle-cell anemia must be a disease of the hemoglobin molecule, with sickle-cell hemoglobin having an abnormal structure...”
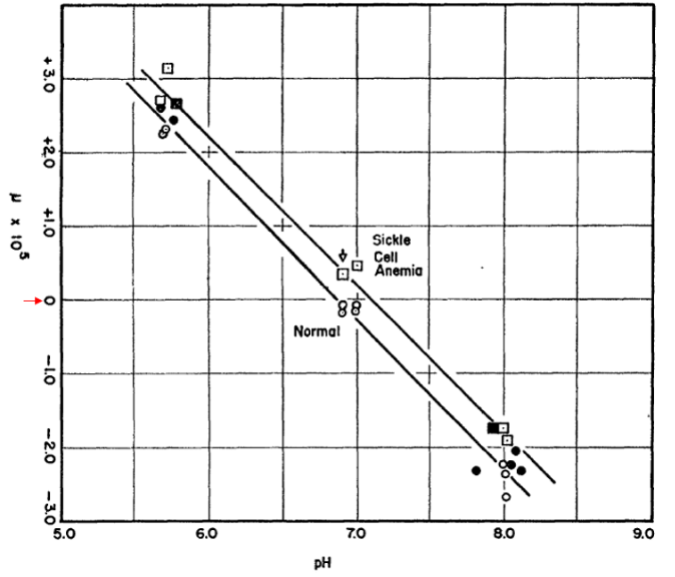
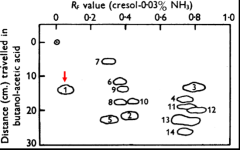
Pauling’s key experiment was to determine the isoelectric points (the pH at which a protein has no net charge) of normal and sickle cell hemoglobin. He did this by measuring the electrophoretic mobility of the proteins over a range of pH values as seen in the figure. Importantly, normal hemoglobin exhibited an isoelectric point of pH 6.87 whereas that of sickle cell hemoglobin was higher, 7.09. This can be seen from the pH (x axis) at which the two proteins exhibited zero mobility as indicated by the 0 (highlighted with a red arrow) in the y axis. This meant that normal hemoglobin has more negative charges than the sickle cell protein. Pauling estimated that sickle cell hemoglobin “has 2-4 more net positive charges” (meaning, 2-4 fewer negative charges) than normal hemoglobin. Later work by Ingram as we will come to showed that the β subunit of sickle cell hemoglobin has a glutamate (acidic) to valine substitution (neutral), meaning that the the sickle cell tetramer, which contains two β-subunits, has two fewer negative charges than the normal protein, which was in close agreement with Pauling’s estimate.
This was an historic discovery because it tied an alteration (mutation) in the genetic material, the gene for the β subunit of hemoglobin on chromosome 11, to a an alteration in a protein. If genes are linear codes that specify proteins, then this was the first demonstration that an alteration to that code resulted in a specific alteration to a particular protein.
Next to enter this story was Vernon Ingram (1924-2006). He was born in Germany but left Nazi Germany when he was 14, settling in England. He studied at the University of London where he obtained a PhD in organic chemistry in 1949. Next, he worked at the Cavendish in Cambridge where in 1952 he did his impactful hemoglobin work. Ingram later (1958) joined the faculty of the Biology Department at MIT.
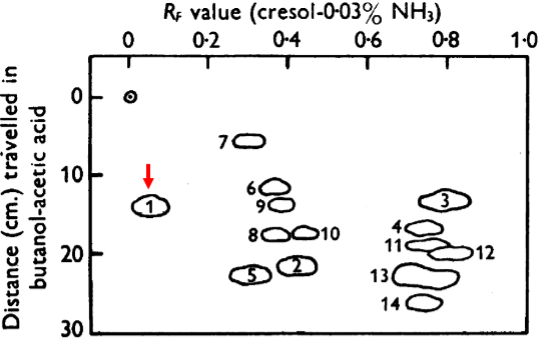
Ingram used two-dimensional chromatography to resolve peptides from hemoglobin that had been generated by treatment with a protease. The peptides were separated by mobility in solvent in one dimension (y axis) and electrophoretically in the x axis where the positive pole was to the right. It can be seen that one peptide exhibited an electrophoretic difference between the two proteins, being more negative in normal hemoglobin than in the sickle cell protein. This peptide is highlighted by the red arrows added to the original figure. Notice that the mobility of this peptide toward the posiitive pole from the sickle cell protein along the x axis lags that of the corresponding peptide from normal hemoglobin. Acidic degradation broke this peptide into smaller fragments, which upon futher analysis revealed that a glutamate, an acidic amino acid, in the normal hemoglobin had been replaced a valine, a neutral amino acid. So, indeed, this was in full accord of the findings of Pauling and further pinpointed the difference to the replacement of glutamate by valine. Ergo, a single mutation resulted in a single amino acid subtitution. (As a footnote to the story, Ingram and another group found an error in and corrected the original published sequence near the site of the amino acid substitution in sickle cell hemoglobin; this correction did not change the principal finding that sickle cell anemia is due to a glutamate to valine substittution.)
In toto, the work of Pauling and Ingram showed that a mutation in a gene could be attributed to a single amino acid substitution in a protien. This was the first direct connection between a gene and its presumed protein product. Complementing the advances of Pauling and Ingram was the elucidation of the complete amino acid sequence of a protein (insulin) by Fred Sanger in work published in 1951 and 1952.
Sanger (1918-2013) did his PhD in the Biochemistry Department at the University of Cambridge in 1943, studying the metabolism of lysine. He then turned his attention to sequencing insulin, work for which he would win the Nobel Prize in Chemistry in 1958. He later moved to the Laboratory of Molecular Biology in Cambridge, which opened in 1962, where he tackled RNA sequencing, coming in second to Robert Holley of Cornell. His second major achievement was developing methods to sequence DNA (chapter 20) for which he was recognized with a second Nobel Prize in Chemistry in 1980 along with Wally Gilbert in 1980.
Insulin was discovered in 1921 by Canadian surgeon Frederick Banting and his assistant Charles Best as a factor that could treat “sugar disease” (diabetes) in children. They isolated the factor from the “Islets [islands] of Langerhans” in the pancreas of a dog. The factor came to be known as insulin from the Latin word insula for island. Its small size and availability as a pure protein in pharmacies made it an attractive candidate for determining the sequence of a protein.
Sanger broke down insulin into peptides using various proteases and partial acid hydrolysis. He then separated the peptides by electrophoresis and chromatography. Next, he subjected the peptides to complete acid hydrolysis so that he could determine their amino acid composition. He also developed a chemical reagent that exhibits a yellow color to label N-terminal amino acids. By breaking down insulin into a variety of different peptides with different proteases, he could assemble the complete, 51-amino acid sequence. For example, the figure shows peptides generated by the protease pepsin that were separated by two-dimensional electrophoresis. Spot 1, which is highlighted with the red arrow, was broken down into the dipeptides glutamate-alanine and valine-glutamate and a tripeptide composed of glutamate, alanine and valine. This enabled him to deduce that the sequence of the Spot 1 peptide was valine-glutamate-alanine.
Sanger discovered that insulin consists of two polypeptide chains (A and B in the figure), which are held together by disulphide bonds (labeled in red) between cysteines. The valine-glutamate-alanine peptide of Spot 1 is highlighted with a blue box. Each chain had a precise amino acid sequence, suggesting by extension that all proteins consist of distinctive sequences of amino acids. This helped set the stage for the Sequence Hypothesis (chapter 24), which held that the order of amino acids in proteins is dictated by the sequences of bases in DNA. In toto, pioneering studies on two proteins -hemoglobin and insulin- helped build a conceptual bridge from genes to proteins. In the video, Sanger, who had a shy, modest personality, explains how he came to discover that insulin consists of two polypeptide chains.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}